
Group Analysis
We often want to generalize the results from our experiment to out of sample data. Here we will infer from the individual subjects activation maps to one activation map for all subjects.
To do this the following steps must be done:
1. Normalize the subjects data to a common space
2. Build a second level GLM
3. Hypothesis testing
The second level GLM has the form
where:
- new design matrix
- estimated from 1st level
Then we can find and perform hypothesis testing on it
Normalize data to MNI template
We will take the computed 1st-level contrasts from the previous experiment and normalize them into MNI-space.
Preparation
We first need to download the already computed deformation field.
Alternatively: Prepare yourself
We're using the precomputed warp field from fmriprep, as this step otherwise would take up too much time. If you're nonetheless interested in computing the warp parameters with ANTs yourself, without using fmriprep, either check out the script ANTS_registration.py or even quicker, use RegistrationSynQuick, Nipype's implementation of antsRegistrationSynQuick.sh.
Normalization with ANTs
The normalization with ANTs requires that you first compute the transformation matrix that would bring the anatomical images of each subject into template space. Depending on your system this might take a few hours per subject. To facilitate this step, the transformation matrix is already computed for the T1 images.
The data for it can be found under:
Now, let's start with the ANTs normalization workflow!
Imports
First, we need to import all the modules we later want to use.
Experiment parameters
We will run the group analysis without subject sub-01, sub-06 and sub-10 because they are left-handed.
This is because all subjects were asked to use their dominant hand, either right or left. There were three subjects (sub-01, sub-06 and sub-10) that were left-handed.
Because of this, We will use only right-handed subjects for the following anlysis.
Specify Nodes
Initiate ANTs interface
Specify input & output stream
Specify where the input data can be found & where and how to save the output data.
Specify Workflow (ANTs)
Create a workflow and connect the interface nodes and the I/O stream to each other.
Visualize the workflow
Run the Workflow
Now that everything is ready, we can run the ANTs normalization workflow.
Visualize Results
First, let's chek the normalization of anatomical image:
And what about the contrast images for Foot > others?
2nd level analysis
After we normalized the subjects data into template space, we can now do the group analysis.
Keep in mind, that the group analysis was only done on N=7 subjects, and that we chose a voxel-wise threshold of p<0.005. Nonetheless, we corrected for multiple comparisons with a cluster-wise FDR threshold of p<0.05.
So let's first look at the contrast average:
Let's take a look at the files we produced
Now, let's see other contrast Foot > others using the glass brain plotting method.
Multiple comparison problem
When we make statistical maps we set a threshold at a given confidence level and declare all voxels for which beta is above this this level as active. Typically our is that the voxel is not active.
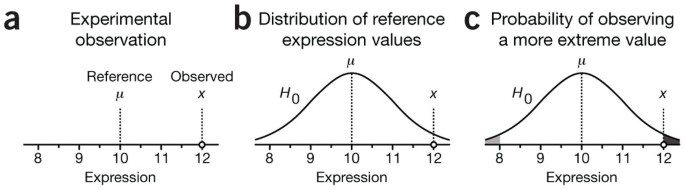
The probability that a test will correctly reject is called the power of the test.
During fMRI experiments we perform test for each voxel, which means there will be many false active voxels. There are many ways to deal with this problem.
How to choose the threshold ?
FWER (family-wize error rate) methods
We control for any false positives : is that there is no activation in any of the voxels.
Control for type 1 errors(We wrongly have rejected the ).
Bonferoni correction. The threshold is adjusted as .
Decreases too much the probability of correctly rejecting .
The voxels are not completely independent.
Random field theory estimates the number of independent statistical tests based upon the spatial correlation, or smoothness.
The number of independent comparisons for smoothed data is:
where -full width at half maximum
At a smoothness of 3 voxels, there would be 1 /27 as many independent comparisons.
The Euler characteristic of the data give us estimation how many clusters of activity should be found by chance at a given statistical threshold
FWER methods often give too conservative threshold, oftentimes FDR gives a better threshold.
FDR (False discovery rate)
Control for the false positives among all declared positives .
Let's take a look how the popular Benjamini and Hochberg FDR procedure work.
Special thanks to Michael Notter for the wonderful nipype tutorial