
fMRI data preprocessing - Introduction
![]()
fMRI scans are saved in dicom format. For scientific analysis of brain images the nifty format (.nii files) are often used. The conversion from dicom to nifty can be done with dcm2niix
Many file are generated during fMRI sessions. These can arranged in many ways, thus a standard is needed how to arrange them. Commonly used standard is Brain Imaging Data Structure (BIDS).
You can use HeuDiConv or Dcm2Bids to automate the conversion from dicom to BIDS.

Let's download the data we will be working with. We will download data through DataLad. It's destibuted data managements system, its provide data storage and version control.
about the data, that we working with: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3641991/
With nibabel we can load a file and inspect its properties.
Lets stop on SliceTiming key, unlike a photograph, in which the entire picture is taken in a single moment, an fMRI volume is acquired in slices. Each of these slices takes time to acquire - from tens to hundreds of milliseconds.
The two most commonly used methods for creating volumes are sequential and interleaved slice acquisition. Sequential slice acquisition acquires each adjacent slice consecutively, either bottom-to-top or top-to-bottom. Interleaved slice acquisition acquires every other slice, and then fills in the gaps on the second pass. Both of these methods are illustrated in the video below.
https://www.brainvoyager.com/bv/doc/UsersGuide/Preprocessing/SliceScanTimeCorrection.html

Introduction Nipype
Why nipype?
Nipype allows to build preprocessing pipelines from different softwares, and it is computationally efficient. There are some helpful ready to use pipleines written with Nipype like fmriprep. To use fmriprep the data have to be in valid BIDS format. The user have to supply only the path to the data setup the parametars.
In Nipype, interfaces are python modules that allow you to use various external packages (e.g. FSL, SPM or FreeSurfer), even if they themselves are written in another programming language than python. Such an interface knows what sort of options an external program has and how to execute it.
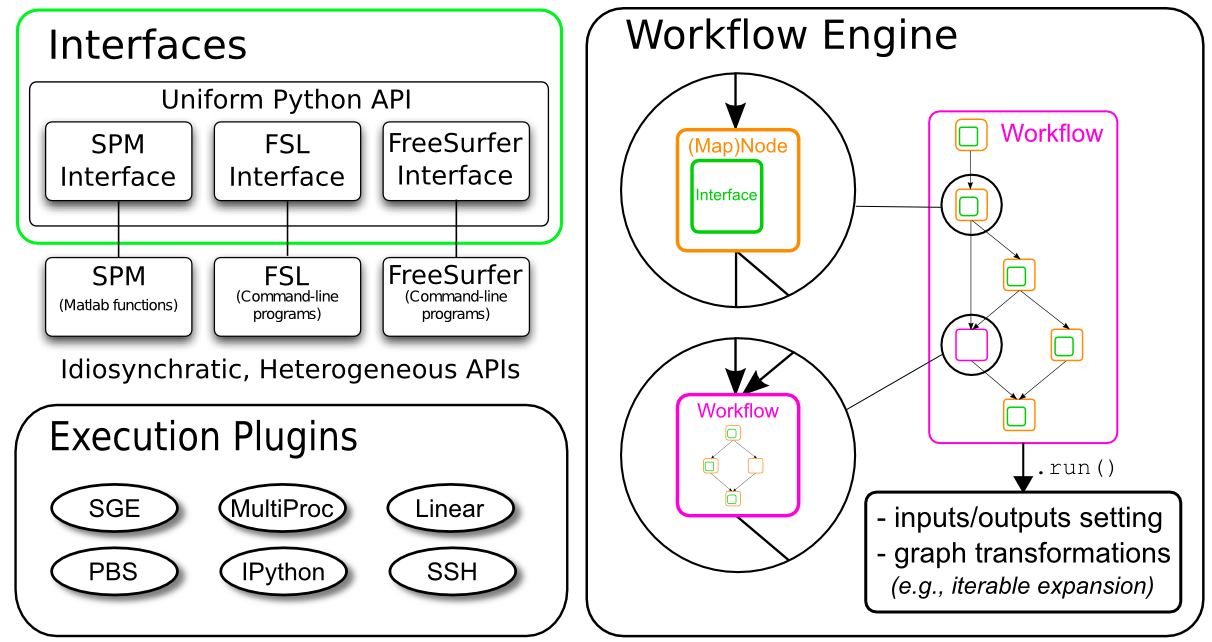
In Nipype, a node is an object that executes a certain function. This function can be anything from a Nipype interface to a user-specified function or an external script. Each node consists of a name, an interface category and at least one input field, and at least one output field.
Once you connect multiple nodes to each other, you create a directed graph. In Nipype we call such graphs either workflows or pipelines. Directed connections can only be established from an output field of a node to an input field of another node.
Dataset: A test-retest fMRI dataset for motor, language and spatial attention functions
About nipype https://www.frontiersin.org/articles/10.3389/fninf.2011.00013/full
Thanks to Michael Notter for the wonderful nipype tutorial